Cannabinoids inhibit neurodegeneration in models of multiple sclerosis
Gareth Pryce*,1, Zubair Ahmed*,1, Deborah J. R. Hankey*,1, Samuel J. Jackson1, J. Ludovic Croxford1, Jennifer M. Pocock1, Catherine Ledent2, Axel Petzold1, Alan J. Thompson3, Gavin Giovannoni1, M. Louise Cuzner1 and David Baker1
1 Department of Neuroinflammation, Institute of Neurology, University College London, London, UK, 2 Institut de Recherche Interdisciplinaire en Biologie Humaine et Moléculaire, Universite libre de Bruxelles, Brussels, Belgium and 3 Neurorehabilitation Group, Institute of Neurology, University College London, Queen Square, London, UK *These authors contributed equally to this work
Correspondence to: Dr David Baker, Institute of Neurology, University College London, 1 Wakefield Street, London WC1N 1PJ, UK E-mail: d.baker@ion.ucl.ac.uk
Received February 19, 2003. Revised April 23, 2003. Accepted April 28, 2003.
Summary
Multiple sclerosis is increasingly being recognized as a neurodegenerative disease that is triggered by inflammatory attack of the CNS. As yet there is no satisfactory treatment. Using experimental allergic encephalo myelitis (EAE), an animal model of multiple sclerosis, we demonstrate that the cannabinoid system is neuroprotective during EAE. Mice deficient in the cannabinoid receptor CB1 tolerate inflammatory and excito toxic insults poorly and develop substantial neurodegeneration following immune attack in EAE. In addition, exogenous CB1 agonists can provide significant neuroprotection from the consequences of inflammatory CNS disease in an experimental allergic uveitis model. Therefore, in addition to symptom management, cannabis may also slow the neurodegenerative processes that ultimately lead to chronic disability in multiple sclerosis and probably other diseases.
Keywords: cannabinoids; excitotoxicity; experimental allergic encephalomyelitis; multiple sclerosis; neuroprotection
Abbreviations: 2-AG = 2-arachidonoyl glycerol; CB = cannabinoid receptor; CREAE = chronic relapsing experimental allergic encephalomyelitis; EAE = experimental allergic encephalomyelitis; EAU = experimental allergic uveitis; ELISA = enzyme-linked immunosorbent assay; i.p. = intraperitoneal; IRBP = interphotoreceptor retinoid binding protein; NMDA = N-methyl-D-aspartate; 9-THC = tetrahydrocannabinol
Introduction
Multiple sclerosis is a chronic disease of the CNS, where autoimmunity is thought to drive the development of inflammatory lesions that induce the primary demyelination, which results in the inhibition of normal neurotransmission (Compston and Coles, 2002). However, the observation that disability often continues to worsen despite immunotherapy, which reduces blood–brain barrier dysfunction and relapse rate (Coles et al., 1999; SPECTRIMS Study Group, 2001; Wiendl and Hohlfeld, 2002) underscores that neurodegenerative changes are of major importance in disease progression (Barnes et al., 1991; Ferguson et al., 1997; Trapp et al., 1998; Coles et al., 1999). This correlates with gross atrophy of the CNS, axonal loss and the accumulation of permanent disability (Bjartmar et al., 2000; Compston and Coles, 2002). Axonal pathology is an early feature of multiple sclerosis lesions and is initially associated with inflammation (De Stefano et al., 2001; Filippi et al., 2003); likewise, axonal damage is a feature in experimental allergic encephalomyelitis (EAE), an autoimmune model of multiple sclerosis (Baker et al., 1990; Wujek et al., 2002). During multiple sclerosis and EAE, destruction of myelin results in the redistribution and aberrant expression of axonal ion channels, and demyelinated axons are particularly sensitive to the damaging effects of free-radicals and glutamate excitotoxicity, which may additionally contribute to chronic neurodegeneration in CNS autoimmune disease (Foster et al., 1980; Black et al., 2000; Pitt et al., 2000; Smith et al., 2000, 2001; Werner et al., 2001; Lo et al., 2002; Kapoor et al., 2003). Therapeutic strategies in multiple sclerosis have concentrated on immunomodulation (Wiendl and Hohlfield, 2002). There is an urgent need for agents that can inhibit progressive multiple sclerosis.
Cannabis contains many compounds but it has been found that the major psychoactive ingredient is 9-tetrahydrocannabinol (9-THC) (Mechoulam and Gaoni, 1967). 9-THC mediates the majority of its activities through stimulation of cannabinoid receptors (CB), notably CB1, which are expressed throughout the CNS (Matsuda et al., 1990; Howlett et al., 2002). Following the discovery of the receptors, fatty acid endogenous ligands, such as anandamide and 2-arachidonoyl glycerol (2-AG), and a degradation system including a re-uptake mechanism and hydrolytic enzymes have been identified (Devane et al., 1992; Deutsch and Chin, 1993; Mechoulam et al., 1995; Dinh et al., 2002). The cannabinoid system functions to regulate synaptic neurotransmission (Kreitzer and Regehr, 2001; Wilson and Nicoll, 2001) and tonically controls clinical signs such as spasticity and tremor that develop in chronic EAE (Baker et al., 2000, 2001). This provides objective evidence to support the claims of multiple sclerosis patients that cannabis may have a benefit in symptom management (Consroe et al., 1997), a claim further supported by some recent clinical trials of medical cannabis extracts (Killestein et al., 2002; Robson et al., 2002; Vaney et al., 2002). There is in vitro evidence that cannabinoids can also regulate glutamate release, oxidant free radicals and calcium influxes (Twitchell et al., 1997; Hampson et al., 1998; Kreitzer and Regehr, 2001; Howlett et al., 2002), which, in excess, can cause neuronal death in neuroinflammatory disease (Pitt et al., 2000; Smith et al., 2000; Kapoor et al., 2003). The lack of specificity of all available cannabinoid reagents (Howlett et al., 2002) and the potential presence of additional CB-like receptors (Di Marzo et al., 2000; Breivogel et al., 2001; Monory et al., 2002) means that gene-deleted transgenic mice (Ledent et al., 1999; Zimmer et al., 1999) provide powerful tools to definitively investigate the potential role of the cannabinoid system in neuroprotection.
Material and methods
Animals
Biozzi ABH and CB1 gene (Cnr1)-deficient mice were from stock bred at the Institute of Neurology. They were fed RM-1(E) diet and water ad libitum. Congenic (N5) ABH.Cnr1 –/– –/+ and +/+ were generated from CD1.Cnr1–/– knockout mice (Ledent et al., 1999) and screened as described previously (Brooks et al., 2002). B10.RIII mice were purchased from Harlan Olac, Oxford, UK. All experiments were ethically performed according to the UK Animals (Scientific Procedures) Act (1984), under the control of the UK Government, Home Office.
Chemicals
The cannabinoid receptor agonists R(+)-WIN55,212 and CP55,940 were purchased from Tocris (Bristol, UK). N-methyl-D-aspartate (NMDA) and the NMDA receptor antagonist MK-801 were obtained from Sigma (Poole, UK). The CB1 selective antagonist rimonabant (SR141617A; Rinaldi-Carmona et al., 1994) and 9-THC were from the National Institute for Drugs and Abuse (NIDA) drug supply program. These were dissolved in ethanol : cremophor : PBS (1 : 1 : 18) and 0.1–0.2 ml was injected intraperitoneally (i.p.) daily.
Induction of chronic relapsing EAE (CREAE)
Mice were injected subcutaneously in the flank on day 0 and 7 with 1 mg mouse spinal cord homogenate in complete Freund’s adjuvant [60 µg Mycobacterium tuberculosis H37Ra, Mycobacterium butyricum (4 : 1) per injection] on day 0 and 7 (Baker et al., 1990). Clinical disease was assessed daily and scored: 0 = normal, 1 = limp tail, 2 = impaired righting reflex, 3 = paresis of hindlimbs, 4 = complete paralysis of hindlimbs and 5 = moribund/death (O’Neill et al., 1992). The activity of animals was monitored over 5 min in a 27 x 27 cm open-field activity chamber (Brooks et al., 2002). Tissues were either snap-frozen or formaldehyde-fixed for immunohistology (Baker et al., 1990; Ahmed et al., 2002), western blotting for caspase activity (Ahmed et al., 2002) and enzyme-linked immunosorbent assay (ELISA) detection of CNS proteins.
Neurofilament ELISA
Whole spinal cords were homogenized on ice by trituration and sonication in 500 µl of barbitone buffer [11 mM barbital, 63 mM sodium barbital, 1.2 mM EDTA (Sigma)] containing a protease inhibitor cocktail and 4 mM EGTA. Lipids were extracted from the sample by adding di-isopropyl-ether (Sigma) at 1 : 5000 and centrifuging for 5 min at 20 000 g. The supernatant was frozen and stored in aliquots at –70°C, and the total protein was measured using the standard Lowry method. Ninety-six-well microtitre plates (Maxisorp; Nunc, Rochester, NY, USA) were coated overnight at 4°C with the SMI35 coat monoclonal antibody (SMI35; Sternberger Monoclonals Inc., Lutherville, MD, USA) against neurofilament heavy chain diluted in 0.05 M sodium carbonate (pH 9.6). This was followed by a wash step with barbitone buffer containing 5 mM EDTA, 1% bovine serum albumin and 0.05% Tween-20 (Sigma). Non-specific protein binding was blocked by incubation with 1% bovine serum albumin in barbitone buffer for 1 h at room temperature, followed by a wash with wash buffer as above. Spinal cord homogenates were serially diluted to 1 : 10 000 in barbitone buffer containing 5 mM EDTA, and incubated at room temperature for 2 h. After washing, a rabbit polyclonal anti-neurofilament H antibody (N-4142; Sigma), diluted 1 : 1000, was incubated at room temperature for 1 h. Following another wash, horseradish peroxidase-conjugated anti-rabbit immunoglobulin diluted 1 : 1000 was incubated for 1 h at room temperature. The tetramethylbenzidine chromogenic reagent (R & D Systems Europe, Minneapolis, MN, USA) was used, signal development stopped using 1 M phosphoric acid, and the plate read at 450 nm, with a reference reading at 620 nm. The antigen concentration for each sample was calculated from an internal standard curve ranging from 0 to 250 ng/ml (high-performance liquid chromatography-purified bovine neurofilament H; Affiniti Bioreagents, Golden, Colorado, USA). All samples were analysed in duplicate.
Induction of experimental allergic uveitis (EAU)
B10.RIII mice were injected subcutaneously with 25 µg interphotoreceptor retinoid binding protein (IRBP151–181) peptide in complete Freund’s adjuvant (as used in EAE experiments) on day 0 and 7, disease was assessed histologically, by haematoxylin and eosin-stained 5 µm paraffin wax sections, for the level of infiltration (score 0–6) and structural damage (score 0–5), as described previously (Hankey et al., 2001).
Glutamate excitotoxcity
NMDA-induced Ca2+ influx
Cerebellar neurons obtained from 6-day-old wild-type ABH.Cnr1+/+ and CB1 knockout mice were cultured for 9 days in poly-D-lysine coverslips as described previously (Evans and Pocock, 1999). At 36 h and 6 days, 10 µM cytosine arabinoside was added to inhibit non-neuronal proliferation (Evans and Pocock, 1999). Cells were loaded with 5 µM of the fluorescent Ca2+ indicator dye fura-2 acetoxymethyl ester (Calbiochem, Nottingham, UK), prior to ionotropic glutamate receptor stimulation with 100 µM NMDA and subsequent inhibition with the NMDA receptor antagonist, 10 µM MK-801. This concentration of MK-801 is required to give maximal block of imaged NMDA-induced Ca2+ influxes in cerebellar neurons (Pocock and Nicholls, 1998). The CB1 receptor agonist CP55,940 (0.01–5 µM) was added 5 min before imaging commenced and was present throughout the experiment. A 12-bit digital camera acquired images and the output visualized with a Life Science Resources Merlin Imaging system, version 1.8630 (Perkin Elmer Life Science, Cambridge, UK). Data were analysed by calculating the 340/380 nm fluorescence ratios with time.
Kainic acid induced lesion
Mice were deeply anaesthetized with halothane and stereotactically injected unilaterally (bregma 2.5 mm, medial-lateral 1.7 mm and dorsoventral 1.6 mm) with 1.5 nmol of kainic acid in 30 µl of 0.9% saline over 30 s; the injection needle was retained for 1 min to prevent reflux of fluid (Chen and Strickland, 1997).
Statistical analysis
Non-parametric data were assessed using the Mann–Whitney U-test with Minitab software (Coventry, UK), parametric data were assessed using t-tests with Sigmastat software. The group score represents the maximal clinical grade developed by all animals within the group.
Results
Development of chronic paresis in EAE is associated with accumulation of axonal loss
Following actively, spinal cord homogenate-induced CREAE, ABH mice develop a relapsing–remitting disease progression of distinct paralytic disease episodes followed by remission with an increasing residual deficit (Baker et al., 1990, 2000). Although histological axonal damage occurs in the initial acute phase of CREAE in ABH mice, this becomes much more evident, particularly in the spinal cord, following the development of relapsing disease (Baker et al., 1990; Ahmed et al., 2002). Whilst EAE has classically been assessed using a subjectively scored scale of paralysis (Table 1; Fig. 1A) (Baker et al., 1990; Smith et al., 2000; Lo et al., 2002), accumulating residual deficit could be quantitatively demonstrated through assessment of mobility of remission animals in an open-field activity chamber (Fig. 1B). Whilst ABH mice remitted to exhibit more immobility after one episode of paralysis (P < 0.001; clinical score 0.5), movement activity was further reduced (P < 0.001) after three to four episodes (clinical grade 2.5–3), where animals had chronically developed residual hind limb paresis upon recovery from relapsing paralytic episodes (Fig. 1B). This immobility was associated with accumulating axonal loss, which could be quantitatively assessed using a neurofilament ELISA (Fig. 2A), and demonstrated histologically (Fig. 2B and C).
Table 1 CB1-deficient mice are susceptible to the development of EAE
Fig. 1
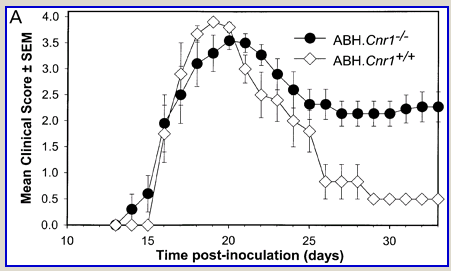
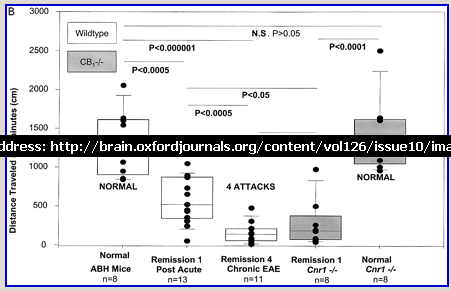
Cannabinoids limit accumulation of disability in EAE. CREAE was actively induced in wild-type ABH or CB1 gene (Cnr1)-deficient, congenic ABH mice with mouse spinal cord homogenate in complete Freund’s adjuvant on day 0 and 7. (A) The mean ± SEM daily clinical scores (post-induction) demonstrate that CB1 knockout mice (filled circles) show poor recovery from paralysis compared with wild-type mice (open diamonds). (B) Movement activity of normal and animals with EAE in remission after one or four paralytic disease episodes were measured in an activity chamber over 5 min. The results represent the individual data points (solid circles) and box plot (25–75% percentile) with 5–95% percentiles range of activity wild-type (open box) and CB1 knockout (shaded box) mice.
Fig. 2
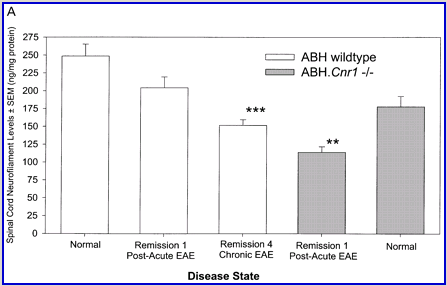
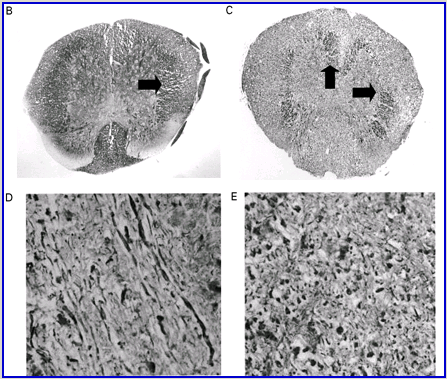
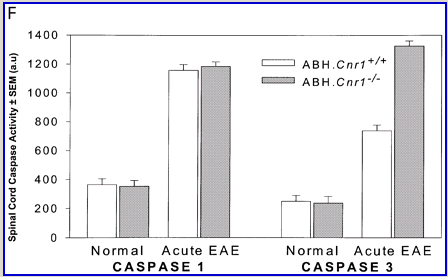
Cannabinoids mediate neuroprotection in experimental allergic encephalomyelitis. CREAE was actively induced in wild-type ABH or CB1 gene (Cnr1)-deficient, congenic ABH mice with mouse spinal cord homogenate in complete Freund’s adjuvant on day 0 and 7, and disease progression in wild-type mice is associated with axonal damage and loss. (A) Spinal cord neurofilament levels from tissue homogenates from wild-type (open boxes) and CB1 knockout (shaded boxes) mice were measured by ELISA from normal and animals with EAE in remission after one or four paralytic disease episodes during EAE. The results represent the mean ± SEM neurofilament levels (n = 6–8 per group). **P < 0.01, ***P < 0.001 compared with respective normal controls. (B–E) Axonal damage was reflected histologically. Bielshowsky silver stain of paraffin 5 µm wax sections of lumbar spinal cord in (B) normal and (C) chronic EAE after four attacks, demonstrating few surviving axons (arrows) and note the loss of axons in the dorsal horn. Neurofilament-specific immunocytochemistry of the spinal cord from a CB1 knockout mouse (D) before and (E) after a single paralytic episode of EAE. Note the many transactions of the white matter axons. (F) Caspase-1 and -3 levels were assessed using western analysis and caspase-3 levels are significantly (P < 0.001) elevated in CB1 knockout mice (shaded boxes) during acute EAE compared with wild type (open boxes) (Student’s t-test; n = 6 per group).
The cannabinoid system regulates EAE-induced neurodegeneration
Wild-type (ABH) and congenic wild-type homozygous (ABH.Cnr1+/+), heterozygotes (ABH.Cnr1+/–) and CB1-deficient (ABH.Cnr1–/–) mice developed EAE with comparable incidence, severity and onset (Table 1). There was, however, an increased frequency (>5%) of EAE-induced mortality in CB1-deficient mice and also in heterozygotes, which have reduced CB1 expression (Selley et al., 2001). However, most strikingly CB1-deficient mice exhibited significantly (P < 0.05) more immobility and residual paresis (Fig. 1) and axonal pathology (Fig. 2A and D–F) than wild-type mice following recovery after the first paralytic episode. These animals relapse and accumulate more deficits that rapidly reach an acceptable severity limit, including the development of permanent hindlimb paralysis. Consistent with the enhanced neurodegeneration after a single attack, spasticity (Baker et al., 2000) developed early in ABH.Cnr1–/– mice, which in wild-type ABH mice usually only occurs after three to four attacks (Baker et al., 2000). Injection of wild-type mice with the CB1 antagonist (twice daily with 5 mg/kg rimonabant i.p. from onset; n = 8) induced greater mortality than usually occurs (<5%). However, using clinical signs of remission, the level of neurodegeneration was not as consistent as that found in CB1-deficient animals (data not shown). Although multiple pathways contribute to axonal damage in EAE, the final effector mechanism in neuronal death is probably toxic ion influxes (Ca2+) and caspase-3-mediated apoptosis (Ahmed et al., 2002), and consistent with this, ABH.Cnr1–/– mice exhibited significantly (P < 0.001) elevated levels of active caspase-3 during acute-phase EAE compared with wild-type ABH.Cnr1–/– mice, although levels of caspase-1 activity were comparable (Fig. 2F). Caspase-3 could be detected immunocytochemically in dying axons and these axons demonstrated many transections, which is a feature of multiple sclerosis (Trapp et al., 1998) (Fig. 2D and E). Therefore, neurodegeneration is clearly elevated in CB1-deficient mice following inflammatory insults, suggesting that CB1 agonism should have neuroprotective potential in CB1-wild-type animals, in addition to controlling neurological symptoms such as tremor and spasticity (Baker et al., 2000).
Exogenous CB1 agonism is neuroprotective in inflammatory CNS disease
In CREAE in ABH mice, the neurological deficit accumulates slowly over a number of months and multi-focal lesions can occur anywhere along the neuroaxis (Baker et al., 1990), complicating assessment and treatment of neurodegeneration, especially as limited quantities of 9-THC were available for study. In contrast, neurodegeneration is restricted to a focal site and develops rapidly in EAU. Following sensitization of B10.RIII mice with IRBP153–180 peptide, the neuroretina is almost completely destroyed within 14–16 days (Hankey et al., 2001) (Fig. 3A–E), again associated with caspase-3-induced neuronal pathology (not shown). CB1 receptor agonism with either R(+)-WIN-55,212-2 (Figs 3C and 4A), at doses that demonstrated no immunosuppressive effect in EAE (vehicle: n = 9/9, clinical score 3.3 ± 0.4, day of onset 17.6 ± 1.2; compared with 5 mg/kg i.p. R(+)-WIN 55,212-2 from day 10–22, n = 7/8, clinical score 3.4 ± 0.5, day of onset 17.3 ± 1.8), and 9-THC significantly inhibited photoreceptor damage, without any apparent inhibition of inflammatory infiltrate (Figs 3E and 4B). Therefore, CB1 agonism can mediate neuroprotection during inflammatory insults.
Fig. 3
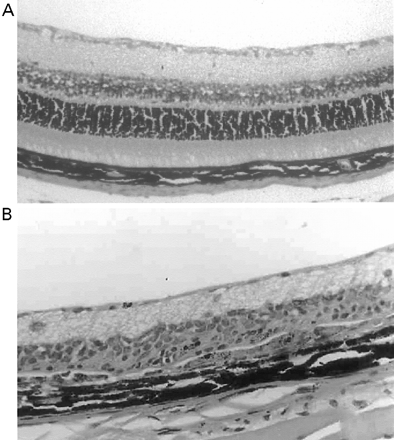
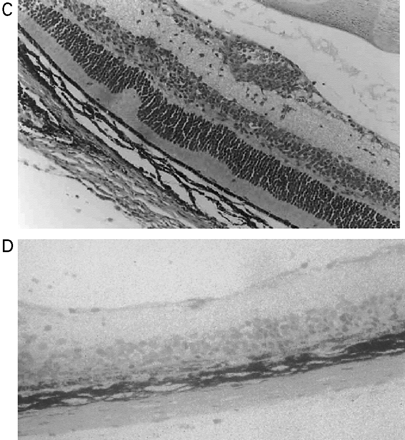
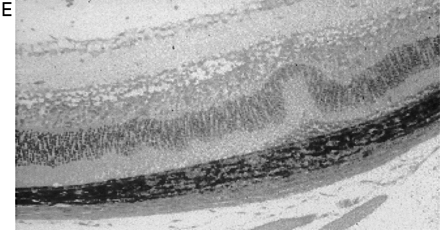
Cannabinoids limit neurodegeneration of the retina in EAU. (A) Paraffin-wax section of normal retina in a B10.RIII mouse. Mice were injected with IRBP153–180 peptide in Freund’s adjuvant and vehicle (B, D) or cannabinoid (C, E) was injected daily from day 8 onwards. Retinas were assessed histologically on either (B, C) day 15 or (D, E) day 21 post-inoculation following treatment with either (C) 5 mg/kg i.p. R(+)-WIN55,212 or (E) 20 mg/kg i.p. 9-THC.
Fig. 4
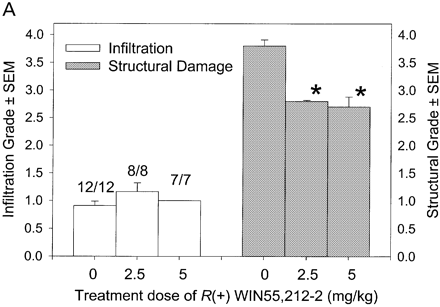
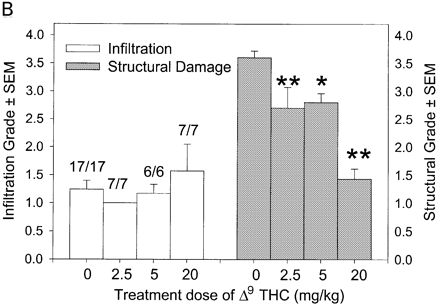
Cannabinoids mediate neuroprotection in EAU. Following active induction of EAU in B10.RIII mice with IRBP153–180 peptide in complete Freund’s adjuvant, eyes were processed for routine wax histology and the level of infiltration (scale 0–5) and structural damage (scale 0–6) assessed. Animals received daily injections of (A) 5 mg/kg i.p. R(+)-WIN55,212 or (B) 20 mg/kg i.p. 9-THC, dissolved in ethanol : cremophor : PBS (1 : 1 : 18), from day 8 onwards. The data represent the results on days 15 and 21 post-inoculation, respectively. The number of animals with EAU per group is indicated. *P < 0.05, **P < 0.01 compared with respective control group.
Cannabinoids regulate/inhibit glutamate excitotoxicity
Although immunosuppression, shown by a reduction in the degree of infiltrate, was not evident following treatment with R(+)-WIN-55, 212–2 or 9-THC (Fig. 3) shortly before expression of disease, 9-THC has been reported to have immunosuppressive effects (Lyman et al., 1989; Wirguin et al., 1994), which could influence neurodegenerative potential. As glutamate excitotoxicity has been implicated in neuronal damage in this and other EAE models (Achiron et al., 2000; Pitt et al., 2000; Smith et al., 2000), glutamate-induced excitotoxicity was examined in vitro and following CNS injection of kainic acid in vivo to examine neuro protective potential in the absence of a compounding immunomodulation. Following in vitro stimulation of NMDA receptors there was a marked (cerebellar) neuronal Ca2+ influx, which was more pronounced in CB1-deficient mice than in controls, suggesting that the cannabinoid system exhibits tonic control of this response (Fig. 5A). In addition, the NMDA receptor antagonist (MK-801) was slower at reducing Ca2+ to basal levels in CB1-deficient mice compared with ABH.Cnr1+/+ wild type, suggesting Ca2+ dysregulation in the absence of CB1 receptors had occurred. Exogenous CB1 agonism by CP55,940 inhibited this NMDA-induced cytosolic Ca2+ influx in wild-type animals, maximally at 1 µM using these culture conditions, but was relatively ineffective in CB1-knockout mice, suggesting that post-synaptic control of NMDA-receptor activation is lost in CB1-deficient mice (Fig. 5A). The injection of kainic acid (>0.15 nM) into CB1-deficient mice induced seizures and caused mortality, usually within 10 min post-injection, which did not occur in wild-type ABH and ABH congenic wild-type (Fig. 5B) mice (P < 0.01), despite using 50-fold higher doses of kainic acid. This elevated mortality was also evident in CD1.Cnr1–/– mice (Fig. 5B), and therefore CB1 receptors are also clearly regulating ionotropic glutamate receptor activity (Fig. 5), which has been implicated in neural exocitoxicity.
Fig. 5
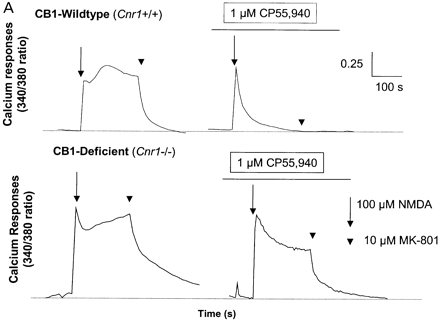
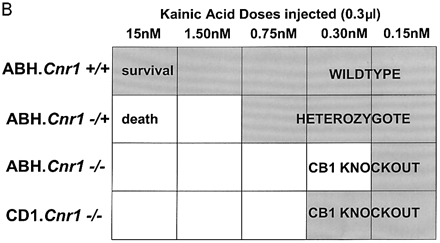
Cannabinoids inhibit glutamate-induced excitotoxicity. (A) Lack of CB1 receptors leads to loss of regulation of NMDA-induced calcium influx. Cerebellar neurons from wild-type and CB1 knockout mice were loaded with the fluorescence Ca2+ indicator dye fura-2 prior to ionotropic glutamate receptor stimulation. Each trace is the mean somatic response of 40 individual neurons measured by single-cell fluorescence imaging. The NMDA receptor agonist, NMDA (100 µM), was added at the arrow and the NMDA receptor antagonist, MK-801, was added at the arrowhead. Where indicated, the CB1 receptor agonist CP55,940 (1 µM) was added 5 min before imaging commenced and was present throughout the experiment. (B) Kainate-induced excitotoxicity in vivo. Halothane anaesthetized animals (n = 5 per group) were intracerebrally injected with 15–0.15 nmol of kainic acid. Rapid (1–10 min) mortality (open blocks) developed in ABH.Cnr1–/– mice compared with the survival (0 out of 5 mortality; shaded blocks) that occurred in wild type (ABH.Cnr1+/+). Seizures consistent with glutamate excitotoxicity were evident. CB1 knockout animals died within 1–3 min of injection, whereas heterozygotes typically showed adverse effects 5–10 min after injection. Doses lower than the maximal survival dose for each strain were not tested
Discussion
Neurological disability in multiple sclerosis correlates with spinal cord axonal loss (50–70% in paralysed multiple sclerosis patients) and reduced N-acetyl aspartate (NAA) levels in chronic multiple sclerosis patients (Bjartmar et al., 2000). This study provides the first and definitive evidence that the cannabinoid system controls the development of neurodegeneration, which occurs as a result of inflammatory insult of the CNS. This provides a novel avenue for neuroprotection in multiple sclerosis and other neurodegenerative diseases.
In diseases such as EAE and multiple sclerosis, it is unlikely that there is a single route to neurodegenerative events, and these may change during the disease course. The clinical outcome will be determined by the rate that these accumulate and how the genetic background of the individual enables them to adapt to the insult. Whilst axonal loss occurs very early in the course of multiple sclerosis (Filippi et al., 2003), it can remain clinically silent for some time, and irreversible neurological disability appears to develop when a threshold (15–30% in mice) of axonal loss is reached and compensatory CNS resources are exhausted (Confavreux et al., 2000; Wujek et al., 2002). In this model, marginal, statistically non-significant axonal loss occurred after the initial neurological attack, assessed here using a novel, relatively rapid neurofilament ELISA and by changes in NAA levels (Preece et al., 1994). Once chronic paresis was evident there was significant axonal loss (40%) as shown here by ELISA and also by magnetic resonance spectroscopy of spinal cord NAA levels (R. A. Page, H. G. Parkes, D. Baker, G. Giovannoni and C. A. Davie, unpublished observations). However, most interestingly, CB1-deficient mice accumulated significant axonal loss (36%) even after a single acute episode, indicating that the presence of CB1 was mediating a degree of neuroprotection during autoimmune attack. Surprisingly, analysis of spinal cord axonal content indicated that apparently normal, CB1-deficient ABH mice have fewer spinal nerves than wild-type animals (P < 0.001), which may be reflective of CB1 involvement in neural plasticity during development (Kim and Thayer, 2001), or there could be inherent neurodegeneration in these animals. This requires further study.
In EAE and, at least initially, in multiple sclerosis axonal damage occurs at least concordantly with inflammation (Ferguson et al., 1997; Trapp et al., 1998), which produces many potentially damaging elements such as cytokines and oxidative stress (Koprowski et al., 1993; Werner et al., 2001; Lock et al., 2002). Ionotropic glutamate receptor systems can also signal damaging mechanisms, at the blood–brain barrier and within the neural microenvironment, in EAE and multiple sclerosis (Bolton and Paul, 1997; Achiron et al., 2000; Pitt et al., 2000; Smith et al., 2000; Kalkers et al., 2002). As shown here, cannabinoids can tonically regulate NMDA glutamate receptor activity in vitro and support the in vivo observation that CB1 regulates NMDA-induced and ischaemic excitotoxicity (Nagayama et al., 1999; Parmentier-Batteur et al., 2002). We also show definitively that CB1 receptor activity regulates kainate glutamate receptor activity in vivo. Cannabinoids also have anti-oxidant properties that could further limit damaging events during inflammation (Hampson et al., 1998; Howlett et al., 2002). In addition, cellular changes such as neural and oligodendrocyte death and gliosis will change the CNS microenvironment, for example through redistribution of ion channels on demyelinated nerves (Foster et al., 1980; Black et al., 2000), loss of trophic support and the formation of compensatory neural pathways, which may contribute to excitotoxic stress and induce further degeneration. This could amplify as the disease progresses, possibly largely independent of inflammation (Compston and Coles, 2002), and may have similarities to nerve destruction in other neurodegenerative conditions, such as Huntington’s chorea, Alzheimer’s disease and amyotrophic lateral sclerosis, where nerve loss accumulates slowly. The cannabinoid system acts as a regulator of many different neurotransmitters and ion (K+ and particularly Ca2+) channels (Henry and Chavkin, 1995; Twitchell et al., 1997; Howlett et al., 2002) and appears to be particularly important when CNS homeostasis is in imbalance, as occurs in disease (Baker et al., 2000). Therefore, CB1 can act at many levels within the death cascade, which will ultimately lead to toxic ion influxes, cell metabolic failure and activation of death effector molecules, such as caspase-3 (Ahmed et al., 2002). This would be consistent with the rapid neurodegeneration that accumulates in CB1-deficient mice. This also implicates a role for endocannabinoids in neuroprotection. The nature of the endogenous neuroprotective cannabinoid has yet to be definitively resolved and may involve more than one CB1-mediated pathway, possibly dependent on the neural circuit involved. Whilst in head trauma it has been suggested that 2-AG may mediate neuroprotection (Panikashvili et al., 2001), in a similar study anandamide, not 2-AG, was shown to be active (Hansen et al., 2001). However, as both anandamide and 2-AG are elevated in chronic EAE lesions (Baker et al., 2001) both may participate in endogenous neuroprotective mechanisms. This will be elucidated once suitable agents to dissect these pathways become available.
Previous studies in non-demyelinating EAE models have demonstrated that high-dose 9-THC, often administered during the induction process, has clinical disease ameliorating effects, due to prevention of infiltrate reaching the CNS (Lyman et al., 1989; Wirguin et al., 1994). Furthermore, 9-THC had no effect on the clinical course, which in Lewis rats is usually naturally self-limiting, when treatment was initiated at disease onset (Lyman et al., 1989). Acute phase paralysis in most rodent EAE models is rapidly reversed and can occur largely independent of any demyelination and axonal loss, and more probably reflects conduction block (Wujek et al., 2002). Therefore, inhibition of acute phase paralysis (Lyman et al., 1989) may reflect inhibition of the immune process that leads to damage in addition to neural effects. Indeed, CB agonists have recently been reported to have immunomodulatory effects in a viral model of multiple sclerosis (Arevalo-Martin et al., 2003; Croxford and Miller, 2003). Without the use of tissue-specific CB1 conditional knockouts (Marsicano et al., 2002), it is probably not possible to completely exclude some influence of cannabinoid inhibition of the immune response in the neurodegenerative process. However, in the context of multiple sclerosis, both would be of benefit for inhibition of disease.
The results of this study are important because they suggest that in addition to symptom management, cannabinoids offer the potential to slow the progression of a disease that as yet has no satisfactory treatment. Therefore, if trials are extended to monitor the long-term effects of cannabis use on symptom management, they should be designed to monitor neuronal loss and progression. If CB1 agonism can be applied whilst limiting the unwanted psychoactive potential, such as through enhancement of endocannabinoid levels (Baker et al., 2001), this may provide a new therapeutic route in multiple sclerosis and could be combined with therapies that target the immunological elements of disease. In neurodegenerative diseases including multiple sclerosis, signs appear once significant damage has already accumulated, slowing the degenerative process early following diagnosis may help improve quality of life for many more years.
Acknowledgements
The Multiple Sclerosis Society of Great Britain and Northern Ireland, the Brain Research Trust, Aims2Cure and the National Institute for Drug Abuse (NIDA) chemical supply program supported this work. Support from the Wellcome Trust and the Alzheimers Trust is also gratefully acknowledged. The authors have declared that they have no conflicting financial interests.
References
Achiron A, Miron S, Lavie V, Margalit R, Biegon A. Dexanabinol (HU-211) effect on experimental autoimmune encephalomyelitis: implications for the treatment of acute relapses of multiple sclerosis. J Neuroimmunol 2000; 102: 26–31.[CrossRef][ISI][Medline]
Ahmed Z, Doward AI, Pryce G, Taylor DL, Pocock JM, Leonard JP, et al. A role for caspase 1 and 3 in the pathology of experimental allergic encephalomyelititis: inflammation verses degeneration. Am J Pathol 2002; 161: 1577–86.[Abstract/Free Full Text]
Arevalo-Martin A, Vela JM, Molina-Holgado E, Borrell J, Guaza C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci 2003; 23: 2511–6.[Abstract/Free Full Text]
Baker D, O’Neill JK, Gschmeissner SE, Wilcox CE, Butter C, Turk JL. Induction of chronic relapsing experimental allergic encephalo myelitis in Biozzi mice. J Neuroimmunol 1990; 28: 261–70.[CrossRef][ISI][Medline]
Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Huffman JW, et al. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature 2000; 404: 84–7.[CrossRef][Medline]
Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Makriyannis A, et al. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J 2001; 15: 300–2.[Free Full Text]
Barnes D, Munro PM, Youl BD, Prineas JW, McDonald WI. The longstanding MS lesion. A quantitative MRI and electron microscopic study. Brain 1991; 114: 1271–80.[Abstract/Free Full Text]
Bjartmar C, Kidd G, Mork S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol 2000; 48: 893–901.[CrossRef][ISI][Medline]
Black JA, Dib-Hajj S, Baker D, Newcombe J, Cuzner ML, Waxman SG. Sensory neuron-specific sodium channel SNS is abnormally expressed in the brains of mice with experimental allergic encephalomyelitis and humans with multiple sclerosis. Proc Natl Acad Sci USA 2000; 97: 11598–602.[Abstract/Free Full Text]
Bolton C, Paul C. MK-801 limits neurovascular dysfunction during experimental allergic encephalomyelitis. J Pharmacol Exp Ther 1997; 282: 397–402.[Abstract/Free Full Text]
Breivogel CS, Griffin G, Di Marzo V, Martin BR. Evidence for a new G protein-coupled cannabinoid receptor in mouse brain. Mol Pharmacol 2001; 60: 155–63.[Abstract/Free Full Text]
Brooks JW, Pryce G, Bisogno T, Jaggar SI, Hankey DJ, Brown P, et al. Arvanil-induced inhibition of spasticity and persistent pain: evidence for therapeutic sites of action different from the vanilloid VR1 receptor and cannabinoid CB1/CB2 receptors. Eur J Pharmacol 2002; 439: 83–92.[CrossRef][ISI][Medline]
Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell 1997; 91: 917–25.[CrossRef][ISI][Medline]
Coles AJ, Wing MG, Molyneux P, Paolillo A, Davie CM, Hale G, et al. Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. Ann Neurol 1999; 46: 296–304.[CrossRef][ISI][Medline]
Compston A, Coles A. Multiple sclerosis. Lancet 2002; 359: 1221–31.[CrossRef][ISI][Medline]
Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis. New Engl J Med 2000; 343: 1430–38.[Abstract/Free Full Text]
Consroe P, Musty R, Rein J, Tillery W, Pertwee R. The perceived effects of smoked cannabis on patients with multiple sclerosis. Eur Neurol 1997; 38: 44–8.[ISI][Medline]
Croxford JL, Miller SD. Immunoregulation of a viral model of multiple sclerosis using the synthetic cannabinoid R(+)WIN55,212. J Clin Invest 2003; 111: 1231–40.[CrossRef][ISI][Medline]
DeStefano N, Narayanan S, Francis GS, Arnaoutelis R, Tartaglia MC, Antel JP, et al. Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol 2001; 58: 65–70.[Abstract/Free Full Text]
Deutsch DG, Chin SA. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol 1993; 46: 791–6.[CrossRef][ISI][Medline]
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992; 258: 1946–9.[Abstract/Free Full Text]
DiMarzo V, Breivogel CS, Tao Q, Bridgen DT, Razdan RK, Zimmer AM, et al. Levels, metabolism, and pharmacological activity of anandamide in CB(1) cannabinoid receptor knockout mice: evidence for non-CB(1), non-CB(2) receptor-mediated actions of anandamide in mouse brain. J Neurochem 2000; 75: 2434–44.[CrossRef][ISI][Medline]
Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA 2002; 99: 10819–24.[Abstract/Free Full Text]
Evans GJO, Pocock JM. Modulation of neurotransmitter release by dihydropyridine-sensitive calcium channels involves tyrosine phosphorylation. Eur J Neurosci 1999; 11: 279–92.[CrossRef][ISI][Medline]
Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain 1997; 120: 393–9.[Abstract/Free Full Text]
Filippi M, Bozzali M, Rovaris M, Gonen O, Kesavadas C, Ghezzi A, et al. Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis. Brain 2003; 126: 433–7.[Abstract/Free Full Text]
Foster RE, Whalen CC, Waxman SG. Reorganization of the axon membrane in demyelinated peripheral nerve fibers: morphological evidence. Science 1980; 210: 661–3.[Abstract/Free Full Text]
Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (-)9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA 1998; 95: 8268–73.[Abstract/Free Full Text]
Hankey DJR, Lightman SL, Baker D. Interphotoreceptor retinoid binding protein peptide-induced uveitis in B10.RIII mice: characterization of disease parameters and immunomodulation. Exp Eye Res 2001; 72: 341–50.[CrossRef][ISI][Medline]
Hansen HH, Schmid PC, Bittigau P, Lastres-Becker I, Berrendero F, Manzanares J, et al. Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem 2001; 78: 1415–27.[CrossRef][ISI][Medline]
Henry DJ, Chavkin C. Activation of inwardly rectifying potassium channels (GIRK1) by co-expressed rat brain cannabinoid receptors in Xenopus oocytes. Neurosci Lett 1995; 186: 91–4.[CrossRef][ISI][Medline]
Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 2002; 54: 161–202.[Abstract/Free Full Text]
Kalkers NF, Barkhof F, Bergers E, van Schijndel R, Polman CH. The effect of the neuroprotective agent riluzole on MRI parameters in primary progressive multiple sclerosis: a pilot study. Mult Scler 2002; 8: 532–3.[Abstract/Free Full Text]
Kapoor R, Davies M, Blaker PA, Hall SM, Smith KJ. Blockers of sodium and calcium entry protect axons from nitric oxide-mediated degeneration. Ann Neurol 2003; 53: 174–80.[CrossRef][ISI][Medline]
Killestein J, Hoogervorst EL, Reif M, Kalkers NF, Van Loenen AC, Staats PG, et al. Safety, tolerability, and efficacy of orally administered cannabinoids in MS. Neurology 2002; 58: 1404–7.[Abstract/Free Full Text]
Kim D, Thayer SA. Cannabinoids inhibit the formation of new synapses between hippocampal neurons in culture. J Neurosci 2001; 21: RC146.[Abstract/Free Full Text]
Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, et al. In vivo expression of inducible nitric oxide synthase in experimentally induced neurologic diseases. Proc Natl Acad Sci USA 1993; 90: 3024–7.[Abstract/Free Full Text]
Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 2001; 29: 717–27.[CrossRef][ISI][Medline]
Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, et al. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 1999; 283: 401–4.[Abstract/Free Full Text]
Lo AC, Black JA, Waxman SG. Neuroprotection of axons with phenytoin in experimental allergic encephalomyelitis. Neuroreport 2002; 13: 1909–12.[CrossRef][ISI][Medline]
Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 2002; 8: 500–8.[CrossRef][ISI][Medline]
Lyman WD, Sonett JR, Brosnan CF, Elkin R, Bornstein MB. Delta 9-tetrahydrocannabinol: a novel treatment for experimental autoimmune encephalomyelitis. J Neuroimmunol 1989; 23: 73–81.[CrossRef][ISI][Medline]
Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature 2002; 418: 530–4.[CrossRef][Medline]
Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990; 346: 561–4.[CrossRef][Medline]
Mechoulam R, Gaoni Y. The absolute configuration of delta-1-tetrahydrocannabinol, the major active constituent of hashish. Tetrahedron Lett 1967; 12: 1109–11.[Medline]
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 1995; 50: 83–90.[CrossRef][ISI][Medline]
Monory K, Tzavara ET, Lexime J, Ledent C, Parmentier M, Borsodi A, et al. Novel, not adenylyl cyclase-coupled cannabinoid binding site in cerebellum of mice. Biochem Biophys Res Commun 2002; 292: 231–5.[CrossRef][ISI][Medline]
Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, et al. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci 1999; 19: 2987–95.[Abstract/Free Full Text]
O’Neill JK, Baker D, Davison AN, Maggon KK, Jaffee BD, Turk JL. Therapy of chronic relapsing experimental allergic encephalomyelitis and the role of the blood-brain barrier: elucidation by the action of Brequinar sodium. J Neuroimmunol 1992; 38: 53–62.[CrossRef][ISI][Medline]
Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, et al. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 2001; 413: 527–31.[CrossRef][Medline]
Parmentier-Batteur S, Jin K, Mao XO, Xie L, Greenberg DA. Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci 2002; 22: 9771–5.[Abstract/Free Full Text]
Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 2000; 6: 67–70.[CrossRef][ISI][Medline]
Pocock JM, Nicholls DG. Exocytotic and nonexocytotic modes of glutamate release from cultured cerebellar granule cells during chemical ischaemia. J Neurochem 1998; 70: 806–13.[ISI][Medline]
Preece NE, Amor S, Baker D, Gadian DG, O’Neill JK, Urenjak J. Experimental encephalomyelitis modulates inositol and taurine in the spinal cord of Biozzi mice. Magn Reson Med 1994; 32: 692–7.[ISI][Medline]
Rinaldi-Carmona M, Barth F, Heaulme M, Shire D, Calandra B, Congy C, et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett 1994; 350: 240–4.[CrossRef][ISI][Medline]
Robson PJ, Wade DT, Makela PM, House H. Cannabis medicinal extracts (CME), including cannabidiol, alleviated neurogenic systems in patients with multiple sclerosis and spinal cord injury. In: 12th Annual Symposium on the Cannabinoids. Burlington (VT): International Cannabinoid Research Society; 2002. p. 56. Available from:www.cannabinoidsociety.org/progab2.pdf. Results of recent phase III trials available from:www.gwpharm.com/news_pres_05_nov_02.html
Selley DE, Rorrer WK, Breivogel CS, Zimmer AM, Zimmer A, Martin BR, et al. Agonist efficacy and receptor efficiency in heterozygous CB1 knockout mice: relationship of reduced CB1 receptor density to G-protein activation. J Neurochem 2001; 77: 1048–57.[CrossRef][ISI][Medline]
Smith KJ, Kapoor R, Hall SM, Davies M. Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol 2001; 49: 470–6.[CrossRef][ISI][Medline]
Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med 2000; 6: 62–6.[CrossRef][ISI][Medline]
60 SPECTRIMS Study Group. Randomized controlled trial of interferon-beta-1a in secondary progressive MS: clinical results. Neurology 2001; 56: 1496–504.[Abstract/Free Full Text]
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. New Engl J Med 1998; 338: 278–85.[Abstract/Free Full Text]
Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol 1997; 78: 43–50.[Abstract/Free Full Text]
Vaney C, Jobin P, Tscopp F, Heinzel M, Schnelle M. Efficacy. Safety and tolerability of an orally administered cannabis extract in the treatment of spasticity in patients with multiple sclerosis. In: 12th Annual Symposium on the Cannabinoids. Burlington (VT): International Cannabinoid Research Society; 2002. p. 57. Available from:www.cannabinoidsociety.org/progab2.pdf
Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol 2001; 50: 169–80.[CrossRef][ISI][Medline]
Wiendl H, Hohlfeld R. Therapeutic approaches in multiple sclerosis: lessons from failed and interrupted treatment trials. BioDrugs 2002; 16: 183–200.[CrossRef][ISI][Medline]
Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 2001; 410: 588–92.[CrossRef][Medline]
Wirguin I, Mechoulam R, Breuer A, Schezen E, Weidenfeld J, Brenner T. Suppression of experimental autoimmune encephalomyelitis by cannabinoids. Immunopharmacology 1994; 28: 209–14.[CrossRef][ISI][Medline]
Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, et al. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol 2002; 61: 23–32.[ISI][Medline]
Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci USA 1999; 96: 5780–85.[Abstract/Free Full Text]










